Process
MRF process

1a: UP000074855_berghei.fasta
1b: result_UP000074855_berghei.csv
2a: A list of proteins from MRF output, should include length of the protein, description
3a: annoted_results.csv (file with annotation using pfsearchV3 and a dataset of profiles for each TR)
4a: details_cluster_report.csv
4b: summary_cluster_report.csv
4c: top_clusters.png
5a: aacompresult_UP000074855_berghei.csv; aacompresult_UP000074855_berghei_by_group.csv
6a: Generate a column in the MRF output file with the length group of each TR
7a: data_composition.csv (from Stefany’s script)
SCRIPTS
1: Run MRF for all the proteins in a proteome fasta file
2: Should obtain all protein ids, description, length, no redundancy
3: execute_annotations.py
4: percent_consensus_dbcscan.py
5: get_aa_composition_regionsH3.py
6: split_mrf_by_groupH1.py
7: TablesTR_fullSteps.py
TAPASS process
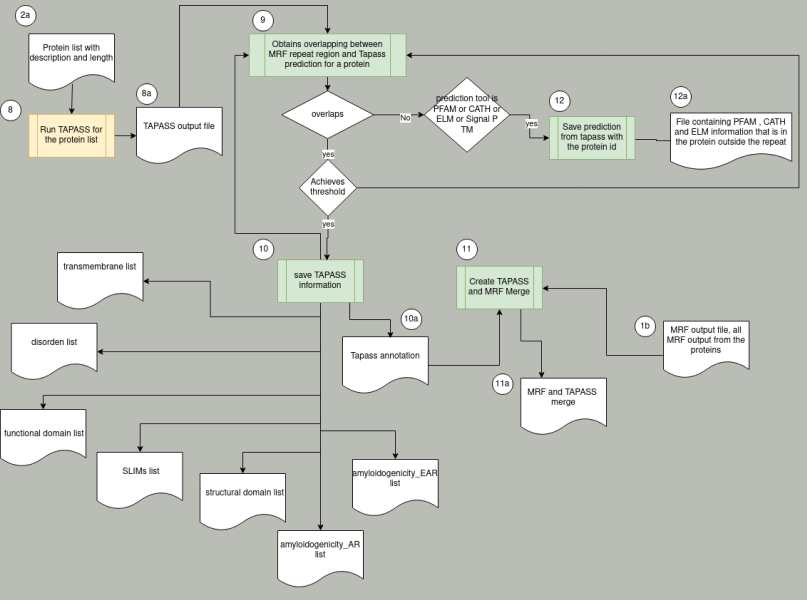
2a: A list of proteins from MRF output, should include length of the protein, description and maybe the sequence from MRF process.
8a:Result_TAPASS_berghei.csv
10a Cath_annotation.csv; pfam_annotation.csv, Slims_annotation.csv
11a merge_complete.csv
12a Cath_annotation_outsideTR.csv; pfam_annotation_outsideTR.csv, Slims_annotation_outsideTR.csv
DESCRIPTIONS
8: executes tapass in the chosen set
9: Get all the lines that correspond to one protein, see which one of them overlaps with a repeat region from MRF. Then for each repeat region analyze all the resulting predicted lines from tapass using the threshold rules.10: If the threshold is achieved save the corresponding tapass lines predicted in the 10a file
11: Create a merge in which the user can select all or some of the following: transmembrane, disorder, functional domain, SLIMs, structural domain, amyloidogenicity_AR and amyloidogenicity_AR for the MRF repeat regions of a chosen set.
12: If there are lines that do not overlap but their prediction tool is PFAM, CATH or ELM or SignalP then save all the corresponding information from tapass in 12a file